レーザ吸収分光法を用いたDME低温酸化機構に関する素反応研究
The Study of Elementary Reaction in the DME Low Temperature Oxidation using Absorption Spectroscopy
須崎光太郎(Kotaro Suzaki)
1. 序論
内燃機関のさらなる高効率化と排気清浄化が求められる中,ノッキング現象,予混合圧縮着火機関制御といった炭化水素の自着火に関わる問題の解決が求められている.近年では自着火問題の解決に化学反応論的アプローチが重要視され,自着火に影響を及ぼすとされる炭化水素の低温酸化過程(T < 1000 K)の詳細な反応メカニズムを明らかにすることが必要となっている.
低温酸化反応は一般的に炭化水素から水素原子が引き抜かれたアルキルラジカル(R)と酸素分子の再結合反応によりアルキルペルオキシラジカル(RO2)が生成する反応により始まる.その後,RO2の内部異性化と酸素第2付加の経路から生成するOHの再生が,消費を上回ることから連鎖的に反応が進むとされる.その一方,温度の上昇に伴いHO2の生成も重要になり,HO2の自己消費反応により低温酸化過程中に蓄積されるH2O2が自着火を引き起こす.HO2は低温酸化過程から自着火への遷移期間において重要な役割を果たす化学種であり,低温酸化過程の進行においてはHO2生成経路と反応を促進させるOH生成経路への分岐比が重要な要素となる.
HO2の生成経路に関して,近年近赤外領域におけるHO2の選択的高感度検出の実現により炭素数の少ないエチル,プロピルラジカルと酸素分子の反応については明らかになった[1, 2].それらによるとHO2の生成過程はRとO2の反応からRO2に蓄積することなく,見かけ上直接的に生成するもの(R1)と,一旦RO2で蓄積し,RO2の熱分解反応により生成するもの(R2)が存在することが明らかになった.
R
+ O2 → HO2
+ alkene (R1)
R
+ O2 ⇔ RO2
→ HO2 + alkene (R2)
また,OHの生成経路に関してもLIFによるOH観測から,反応開始直後に生成するものが存在することが明らかになり,RとO2から(R1)同様RO2で蓄積せず見かけ上直接生成するものがあることが示された[3].
R
+ O2 → OH +
o-hetelocycle (R3)
本研究では炭化水素に酸素原子を含むエーテル系で最も単純なジメチルエーテル(DME,CH3OCH3)を用いた.DMEは自着火性が高く,次世代の燃料の一つとしても検討されている.DMEの低温酸化反応はメトキシメチルペルオキシラジカル(CH3OCH2O2)の内部異性化を経由したOHの生成が有利で,OHの再生による連鎖反応であることがモデルの上では示されているが,素反応レベルでは検証されていなかった[4].HO2の生成経路についてもこれまで実験的には全く検討されていなかった.
そこで本研究ではHO2,OHを近赤外領域により観測し,さらに同一のセルにおいてRO2を紫外吸収分光法により観測することからDME低温酸化過程を素反応レベルで検討し,HO2の生成経路を明らかにすることを目的とした.
2. 実験手法
反応はNd-YAGレーザの3倍高調波(355 nm)によるDME / O2
/ Cl2 混合気体への閃光分解法を用い,酸素濃度を過剰にすることで,塩素分子の2次反応が無視できる条件で行った.
Cl2
+ hν → 2 Cl (R4)
Cl
+ CH3OCH3 → CH3OCH2 + HCl (R5)
CH3OCH2
+ O2 → products (R6)
HO2の初期塩素原子濃度に対する生成割合はCH3OH / O2
/ Cl2の混合気体から得られる信号を参照して得た.
Cl2
+ hν → 2 Cl (R4)
Cl
+ CH3OH → CH2OH
+ HCl (R7)
CH2OH
+ O2 → HO2
+ HCHO (R8)
初期塩素濃度に対して十分なCH3OHがあれば(R7),(R8)は100%進行することから,生成するHO2は初期塩素原子濃度と等しい.そのため初期塩素原子濃度が等しい条件において(R8)で生成したHO2シグナルに対して(R6)で生成するHO2シグナルを比較することにより初期塩素原子濃度に対する生成割合(収率)を得ることができる.OHは上記混合気体に NOを加えることによりOH参照シグナルを得た.
HO2
+ NO → OH + NO2 (R9)
温度は298 Kから625 Kまで,圧力を20 torrから90 torrまで変化させた.典型的な実験条件は[Cl2] = 2×1014,[O2] = 1.2×1016,[CH3OCH3] = 1×1015 molecule cm-3とし,このとき,初期塩素原子濃度はおよそ1×1013 molecule cm-3であった.
観測はHO2,OHを近赤外周波数変調分光法とHerriott型長光路吸収セルを用いて観測し,RO2はそれらに干渉することなく同じセルに設置した紫外吸収光学系により観測した.装置概要をFig. 1に示し,それぞれの観測方法を説明する.
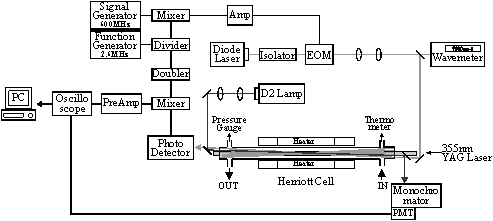
Fig. 1 Experimental apparatus
2.1 近赤外周波数変調分光法とHerriott型セル
1.4 μmのダイオードレーザから発した近赤外光はElectro Optic Modulator(EOM)により600 ± 2.6 MHzに変調され,Herriott型のセルを透過した後,フォトディテクターにより検出される.位相変調を受けた搬送波とサイドバンドとの吸収強度差により,ディテクターで検出されたシグナルには吸収量に比例したビートが現れる.副変調周波数の2倍の周波数成分(5.2
MHz)であるそのビート信号を検波回路により取り出し,デジタルオシロスコープで積算し,シグナルを取り込んだ.反応容器は内径が41 mmの石英管からなり,多重反射凹面鏡対を間隔約1500 mmとして真空槽内に設置した.検出用レーザの反射回数は24回で,分解光と検出光の重なる領域が約60 cmであるので,有効光路長は約15 mになる.容器中心部はヒータで覆われており,800 Kまで温度調節が可能になっている.HO2は第一電子励起状態への遷移帯にある7013.520 cm-1 (Q2(18)1-0),OHはO – Hの振動倍音吸収帯の6971.291 cm-1 (Q1e(1.5))にて観測した.これらはダイオードレーザ1素子の可変波長域内にある.
2.2 紫外吸収分光法
CH3OCH2O2の観測を行うため,上述した同じセルに紫外吸収光学系を構成した.光源に重水素ランプを用い,2枚のレンズでコリメートした紫外光を,同じセル内に,近赤外光が出射する部分から入射した.セル内を透過した紫外光を,分光器に入射し,光電子増倍管により検出した.CH3OCH2O2は250 nmで観測し,典型的な条件での見かけの吸光度はおよそ4×10-3 である.この波長ではHO2の吸収帯も重なるため,近赤外領域で得られたHO2信号を用いて補正を行った.
3. 結果及び考察
3.1 生成物観測
Figure 2に600 Kと298 KにおけるHO2,OH,CH3OCH2O2の時間プロファイルを示した.時間プロファイルは初期塩素濃度に対する割合で表している.600 KではHO2は徐々に生成し,OHは反応開始直後に急激に生成し,いずれもその後減衰する.OHの収率は直鎖の炭化水素での例に比べおよそ20倍であり[3],OH生成経路がDMEではより有利であることを示唆している.CH3OCH2O2は反応開始後に急激に生成し,その後減衰して時間変化がなくなるという結果を得た.298 KではHO2は反応開始直後に微量生成し,その後減衰せず一定のシグナルを示した.OHは298 Kにおいても生成し,反応開始直後に生成し,その後減衰した.また,CH3OCH2O2は反応開始直後に生成し,およそCH3OCH2O2の自己消費反応の速度定数に従い減衰した.
また,観測した化学種の温度に対する収率の変化をFig. 3に示した.OH及びCH3OCH2O2の収率はピーク値,HO2は自己消費反応による減少分を補正した値である.HO2は500 Kまでは約10%を保ち,500 Kを超えると急激に収率が上昇し約4割となった.OHは温度の上昇に伴い徐々に増加し,600 Kではおよそ4割となった.また,CH3OCH2O2は500 Kまでは徐々に減少し,500 Kを超えると急激に減少した.これはCH3OCH2O2の熱分解反応がこの温度域から始まっていると考えられる.
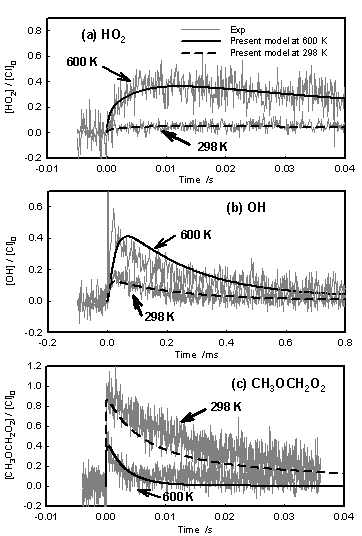
Fig.2 Time profiles of (a) HO2, (b) OH and (c) CH3OCH2O2 at 600 K and 298 K.
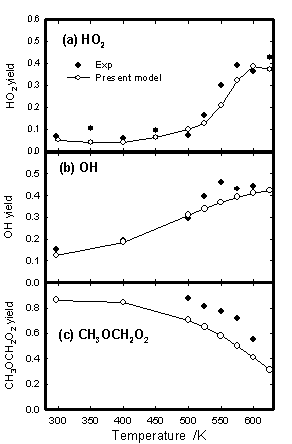
Fig. 3 Yields of (a) HO2, (b) OH and (c) CH3OCH2O2 as a function of temperature
3.2 600 KにおけるHO2生成メカニズム
既存のメカニズム[4]において600 K近辺でのHO2の生成経路は,以下の反応経路で示されている.
CH3OCH2 + O2 ⇔ CH3OCH2O2 (R10)
CH3OCH2O2 ⇔ CH2OCH2OOH (R11)
CH2OCH2OOH → 2 HCHO + OH (R12)
HCHO + OH → HCO + H2O (R13)
HCO + O2 → HO2 + CO (R14)
CH3OCH2 → CH3 + HCHO (R15)
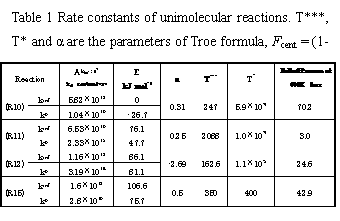
600 Kでは(R10)と(R15)が競合する.ところが既存のメカニズムでは本実験で得られた時間プロファイルは再現されない.既存のモデルは常圧以上の条件に最適化されているため,圧力依存を示す単分子反応の速度定数は高圧極限の値が採用されている.そこで圧力依存を示す(R10) ~ (R12)及び(R15)の速度定数を(R10) ~ (R12)はYamadaらの理論計算値[5]を用いて,(R15)は本実験装置を用いて実測し,Troeの式で表現した.その値をTable 1に示す.また,本実験装置において,紫外吸収分光法により600 Kから700 K,20から90 torrの条件で228 nmにおいてCH3OCH2を観測し,(R15)の速度定数を計測した結果をFig. 4に示した.図中の太線はTroe式を用いた速度定数を示しており,実測値を良く再現している.20から90 torrの圧力条件ではこれらの反応の速度定数はほぼfall–off領域にあることがわかる.しかし,これらの修正のみでは,どの時間変化も再現されない.
そこで,HO2,OHの時間変化を再現するために以下の反応を考慮した.
CH3OCH2
+ O2 → OH + 2 HCHO (R16)
CH3OCH2
+ O2 → HCO + HCHO
+ H2O (R17)
CH3OCH2O2
+ OH → HO2 + CH3OCH2O (R18)
このとき,各速度定数をk16 = 4.0×10-13,k17 = 5.8×10-14,k18 = 4.0 ´ 10-11
cm3 molecule-1 s-1としたとき,各化学種の時間プロファイルはFig. 2に示した通りほぼ再現された.
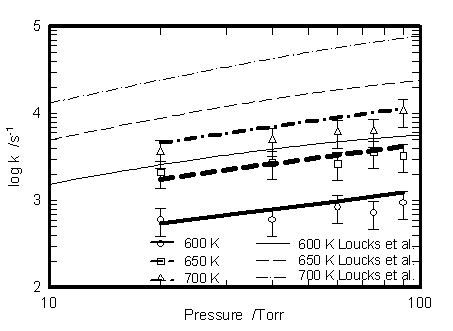
Fig. 4 Rate constant of CH3OCH2 decomposition reaction as a function of pressure. Symbols are measurement results, bold lines are fitting using Troe form expression
Figure 5にG2M(UCC1)法により得たCH3OCH2 + O2反応系のポテンシャルエネルギーダイヤグラムを示す.直鎖の炭化水素では有利な経路として示された,RO2からのHO2生成経路はDMEにおいては反応障壁が高く,本温度域ではその寄与はほぼないと考えられる.本計算により新たにCH2OCH2OOHの内部異性化後,HOCH2OCH2OからHCOが生成する経路が存在することが示された.HCO生成後は (R14)によりHO2が生成する.
以上から,600 KにおいてHO2は主にHCOを経由して生成することが明らかになった.HCOは既存のモデルにおけるHCHO + OHから生成するが,それだけではHO2生成の全てを説明できず,反応開始直後のHO2にはQOOHからHOQOへの内部異性化を経由する新たな反応経路によりCH3OCH2 + O2から直接的に生成するHCOが寄与する可能性があることが示された.またOHも反応開始直後に生成し,CH3OCH2 + O2から直接的に生成してくる経路も必要であることが示された.
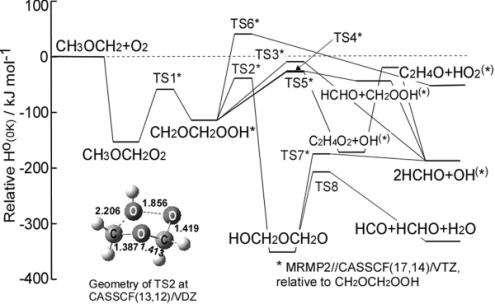
Fig. 5 Potential energy diagram of CH3OCH2 + O2 reaction
3.3 298 KにおけるHO2生成メカニズム
298 Kにおいては既存のメカニズム[6]では以下の反応によりHO2が生成するとされている.
CH3OCH2 + O2 ⇔ CH3OCH2O2 (R10)
CH3OCH2O2 + CH3OCH2O2
→ 2 CH3OCH2O + O2 (R19-a)
→ CH3OCHO + CH3OCH2OH + O2 (R19-b)
CH3OCH2O → CH3OCHO + H (R20)
CH3OCH2O + O2 → HO2 + CH3OCHO (R21)
H + O2 → HO2 (R22)
しかし,既存のモデルだけでは本実験のプロファイルは再現されず,600 K同様に(R16) ~ (R18)の反応を考慮したとき,Fig. 2に示したとおり,3つの化学種のプロファイルを再現することができた.
このとき,(R18)の速度定数は温度に依存しないとし,k18 = 4.0 ´ 10-11 cm3 molecule-1 s-1とした.(R16),(R17)の速度定数は各温度におけるプロファイルから,k16 = 1.55 ´ 10-13 ´ exp( 4.7 kJ / mol / RT), k17 = 1.1 ´ 10-13 ´ exp( - 3.2 kJ / mol / RT ) cm3 molecule-1 s-1とした.Figure 3に示したとおり,各温度における収率を良く再現している.
本温度域においてHO2は,炭素数の少ない炭化水素のようにRO2で蓄積せずに直接的に生成する経路とは異なり,CH3OCH2O2の自己消費反応から始まる一連の反応から生成する.また,反応開始直後のHO2はCH3OCH2 + O2からのHCOの直接生成経路とOH + CH3OCH2O2の反応により生成することが明らかになった.
本温度域でHO2生成に重要な,CH3OCH2O2の自己消費反応の速度定数は,本実験装置を用いて紫外吸収分光法によるCH3OCH2O2の時間プロファイルから決定した.計測温度は298 ~ 400 Kとした.Figure 6に,計測した速度定数のアレニウスプロットを示した.この結果からCH3OCH2O2の自己消費反応の速度定数を
k19 = (3.9 ± 1.9)×10-13×exp(720 ± 150 / T) cm3molecule-1s-1
と得た.
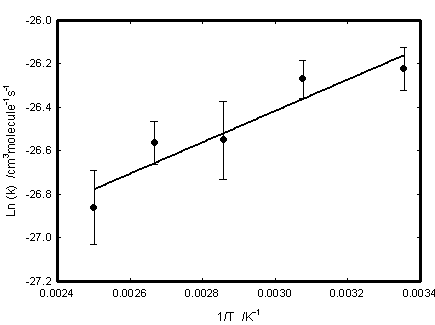
Fig. 6 Arrenius plot of the CH3OCH2O2 self-reaction rate constant between 298 K and 400 K
4. 結論
本研究では近赤外周波数変調分光法とHerriott型長光路吸収セルを用いてHO2及びOHを一つのダイオードレーザの可変波長域内で観測し,また同じセルに設置した紫外吸収光学系によりRO2の観測を行い,低温酸化過程において重要なHO2,OH,RO2を同一条件で観測できる装置を実現した.それらを用い,DMEの低温酸化過程におけるHO2生成経路の検討を行い,HO2は500 K近辺を機構の切替わる境界として,600 K近辺ではHCOを経由する経路から生成し,298 K近辺ではRO2の自己消費反応から主に生成することを明らかにした.また新たにQOOH → HOQOの内部異性化からHCOが生成する経路が量子化学計算においても示され,CH3OCH2 + O2から直接生成するHCOが各温度域において反応開始直後のHO2生成に寄与することが示された.これらからDMEの低温酸化過程におけるHO2は,これまでにHO2生成経路が示されている炭化水素とは異なる経路で生成することが明らかになった.素反応レベルで新たなHO2生成経路を明確にした点において,炭化水素の燃焼モデル構築に適用可能な知見を得ることができた.
参考文献
1. Clifford. E. P, Farrell. J. T, DeSain. J. D, and Taatjes. C. A, J. Phys. Chem. A, 2000. 104. 11549-11560
2. DeSain. J. D, Clifford. E. P, and Taatjes. C. A, J. Phys. Chem. A, 2001. 105. 3205-3213
3. DeSain. J. D, Klippenstein. S. J, and Taatjes. C. A, Phys. Chem. Chem. Phys, 2003. 5. 1584-1592
4. Curran. H. J, Pitz. W. J, Westbrook. C. K, Dagaut. P, Boettner. J. C, and Cathonnet. M, Int. J. Chem. Kinet. 1998. 30. 229-241
5. Yamada. T, Bozzelli. J. W, and Lay. T. H, Int. J. Chem. Kinet. 2000. 32. 435-452
6. Jenkin. M. E, Hayman. G. D, Wallington. T. J, Hurley. M. D, Ball. J. C, Nielsen. O. J, and Ellermann T, J. Phys. Chem. 1993. 97. 11712-11723