炭化水素の低温酸化反応過程に関する素反応のラジカル分光計測
Spectroscopic Analysis of Radical Reactions in Low Temperature Oxidation of Hydrocarbons
佐藤義久(Yoshihisa SATO)
1. 背景
炭化水素ラジカルの素反応機構を解明することは,燃焼現象の理解に対して重要な課題である.本研究では,HCCI機関の制御やエンジンのノッキングの抑制に重要と考えられている低温酸化反応に注目し,レーザ分光を用いて未解明の反応素過程に対して反応速度の測定・生成物の検出と定量を試みた.
本研究で注目した炭化水素ラジカル種は,ビノキシラジカル(CH2CHO)のメチル置換体である1-メチルビノキシラジカル(CH2C(O)CH3)と2-メチルビノキシラジカル(CH3CHCHO),シクロヘキシルラジカル(c-C6H11)である.これらの反応に関して,ラジカル,生成物をレーザ分光計測により観測し,反応速度の測定と生成物の定量を行った.
2.メチルビノキシラジカルと酸素分子の反応
2.1実験手法
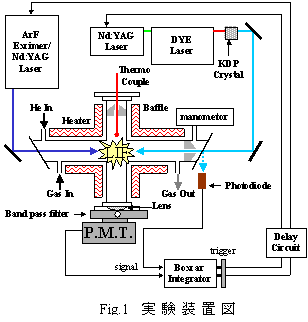
実験は298-700Kの温度範囲においてレーザ光分解レーザ誘起蛍光法(LP-LIF)を用いて行った.実験装置をFig.1に示す.ラジカル前駆体,反応ガス,バッファー(He)はマスフロコントローラにより流量調節し,十分に混合してから反応セルへ導入した.OH収率の測定に用いたNOは,混合中にO2と反応するため,独立した配管でセルへ直接流入させた.全流量は光分解レーザの照射間隔内にセル内のガスが完全に入れ替わるように十分な流量に調節した.
ラジカル前駆体の光分解にはArF (193nm),又はNd:YAGレーザの三倍波(355nm)を用い,OH,CH2C(O)CH3の検出にはそれぞれ,Nd:YAGの倍波(532nm)励起の色素レーザを二倍波とした307.9nmのA-X(0,0)遷移Q1(1)線,341.6nmのA-X遷移kピークにより検出を行った.光分解レーザとプローブ光は同軸直線上に反対側からセル内へ導入した.OHからの蛍光は,装置中央に設置された光電子増倍管により検出し,ボックスカー積分器により10回または30回積算された後PCに記録した.

2.2 CH2C(O)CH3+O2:1-メチルビノキシと酸素の反応
2.2.1 測定に用いた反応系
CH2C(O)CH3はアセトン(CH3C(O)CH3)と塩素原子(Cl)の反応を用いて生成した.O2との反応により減少するCH2C(O)CH3を検出し反応速度定数の測定を行い,また,生成するOHを検出し,その収率の定量を行った.
Cl2 + 355nm → 2Cl
CH3C(O)CH3
+ Cl → CH2C(O)CH3 + HCl
CH2C(O)CH3
+ O2 → OH + Products
OHの定量には,CH3C(O)CH3をメタノール(CH3OH)に置き換え,さらにNOを加えて参照系とした.この反応系において,Cl原子初期濃度と同量のOHが生成する.参照系におけるLIF強度を基準とし,CH2C(O)CH3+O2反応系より生成するOHのLIF強度を比較してOH収率の定量を行った.
Cl2 + 355nm → 2Cl
Cl + CH3OH → CH2OH
+ HCl
CH2OH + O2
→ HO2
+ CH2O
HO2 + NO → OH + NO2
2.2.2 結果・考察
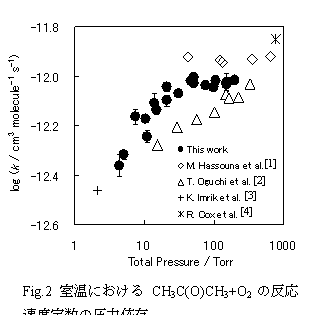
CH2C(O)CH3+O2の反応速度
CH2C(O)CH3+O2の反応速度定数は室温における圧力依存に関して二つの報告が対立している.測定した圧力範囲(31.9-7600 Torr)において,反応速度の圧力依存がないとしたHassouna[1]らに対してOguchi[2]らは15.3-333 Torrにおいて測定を行い,100 Torr付近より速度が減少する傾向を報告した.本研究で測定した反応速度定数はOguchiらよりもfalloff圧力が低圧側へシフトしており,Hassounaらの計測した圧力範囲においては,圧力依存性の効果が弱い傾向を示した (Fig.2).本研究で更に低圧での測定を行った結果,30Torr付近より明確な圧力依存性を示すことが明らかとなった.

OH収率の定量
CH2C(O)CH3+O2の生成物に含まれると考えられるOHの検出を試みたところ,室温においてOHは観測されず,700Kにおいてもわずかに観測されたものの,CH2C(O)CH3+O2からの直接的な生成物であるとは確認出来なかった. 700K, 35Torrにおいて参照系とのシグナル強度の比較によりOH収率の上限値を求めたところφ<0.02を得た.室温,40TorrにおいてOHの収率が0.15のビノキシラジカル(CH2CHO)とは大きく異なり,CH2C(O)CH3+O2より生成するOHの収率は大きく減少する(Fig.2).これより,CH2CHOに対してホルミル基(CHO)のHをメチル(CH3)置換することによりOHの収率は大きく減少すると考えられる.
2.3 CH3CHCHO+O2:2-メチルビノキシと酸素の反応
2.3.1 実験に用いた反応系
CH3CHCHOは1-プロペニルエチルエーテル(CH3CHCHOC2H5)の光分解(193nm)により生成させた.その後,O2との反応により生成するOHをLIFにより検出した.
CH3CHCHOC2H5
+ 193nm → CH3CHCHO + C2H5
CH3CHCHO+O2 → OH + Products
生成したOHはN2Oの光分解により生成する一重項励起状態の酸素原子(O(1D))と水素分子(H2)の反応により生成するOHのLIF強度を基準として定量を行った.
N2O + 193nm → O(1D)
+ N2
O(1D) + H2 → OH + H
実験は [Precursor],[O2]>>[Radical]の成り立つ条件においてOHのLIFシグナルの時間変化を測定した.はじめに,Precursor,O2濃度に対する擬一次反応速度の変化より二次反応速度定数を求め,次に,生成するOH収率の評価を行った.OH-LIFシグナルのO2によるクエンチングはBailey[2]らの評価した係数を用いて補正した.また,O(1D)+H2より生成するOHはv>0の振動状態を取り得るため,NOを加えてv=0へ緩和したシグナルの強度を基準としてOHの収率を評価した.本実験は全て室温で行った.
2.3.2 結果・考察
OH収率の定量CH3CHCHO + O2より生成するOHラジカルの生成速度はOguchi[2]らによって求められた反応速度と一致し,OHラジカルが直接的な生成物であると確認された.10-100Torrの範囲でOH収率を測定したところ,収率は圧力に対して依存し,およそ0.08-0.02となった(Fig.3).OH収率の圧力依存に関して,CH2CHO + O2の場合と同様に,圧力の増加に伴い収率が減少し,再結合生成物への安定化と競合していると考えられる.OHの反応収率がCH2CHOに比べて半分以下に減少したことについては,CH3置換基の付加により分子内自由度が増加し,O2が付加して生成するCH3CH(O2)CHOの有するエネルギーがより多くの反応座標に分散されるため,OH生成へと進む反応座標に対するエネルギーが減少したためだと考えられる.
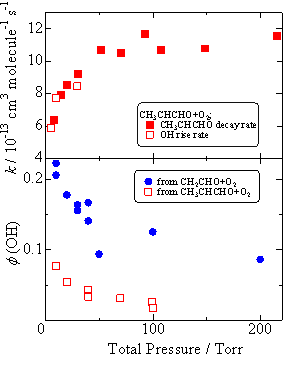
Fig.3 OHの生成速度とCH3CHCHO+O2の反応速度の比較と,OH収率の圧力依存
2.4 まとめ
CH2CHO,CH2C(O)CH3,CH3CHCHOのビノキシ型ラジカルの反応を比較すると,メチル置換基の付加位置により,以下の影響を及ぼす.
・CH3置換基が付加することにより, OH収率が減少し,-CHO基のHがCH3に置換されることでOHの収率は大きく減少する.
・ビノキシ型ラジカルが酸化反応においてOH生成には,-CHO基を有していることが重要である.
3. シクロヘキシルラジカルと酸素分子の反応
3.1 実験
実験装置はこれまで我々がジメチルエーテルの酸化機構の検討に用いたレーザ閃光分解・準静的流通反応管である.298-750Kにおいて近赤外周波数変調(NIR-FM)分光法を用いてHO2,OHの検出を行い,UV吸収法を用いてc-C6H11O2の検出を行った.実験装置図をFig.4に示す.試料気体,バッファー(He)はマスフロコントローラにより流量調整し,反応セルに導入した.また,総流量は光分解レーザの照射(1Hz)毎にセル内のガスが完全に入れ替わるのに十分な流量(4.7SLM)に設定した.
一連のラジカル反応の開始には,パルスNd:YAGレーザ(三倍波: 355nm)を用いてCl2を光分解させ生成したCl原子を用いた.HO2とOHの検出には連続発信チューナブルダイオードレーザを光源として,それぞれA-X(0’-0”)遷移の7013.520cm-1,倍音振動の6971.394cm-1を中心波長とし,多重反射長光路吸収をNIR-FMにより検出した.またc-C6H11O2は,D2ランプを光源としたシングルパス吸収により,253nmを中心波長に持つバンドパスフィルタ(FWHM:10nm)の透過光を用いて検出した.閃光分解レーザパルス照射を起点として時間分解観測されたシグナルは,オシロスコープにより1000回積算し,PCに保存した.


実験に用いた反応系を以下に示す.
Cl2 + 355nm → 2Cl
c-C6H12 + Cl → c-C6H11 + HCl
c-C6H11 + O2 → (c-C6H11O2)* → HO2 + c-C6H10
(c-C6H11O2)* → OH + c-C6H10O
(c-C6H11O2)* (+M) ⇔ c-C6H11O2 (+M)
HO2の定量は,同様にして生成したClを用いてCH3OH,O2との反応により生成するHO2のシグナル強度を基準として行った.OHの定量は生成したHO2をNOと反応させ生成したOHのシグナル強度を基準として行った.
CH3OH + Cl → CH2OH + HCl
CH2OH + O2 → HO2 + CH2O
HO2 + NO → OH + NO2
実験は全て分子総密度[M]=0.2-1.2×1018 molecule cm-3一定,[c-C6H11]及び [CH2OH]<<[O2],[Cl]<<[c-C6H12]及び[CH3OH] の擬一次条件の成り立つ範囲で行った.また、本研究は温度T=298-750Kの範囲で実験を行った.
3.2 結果・考察
c-C6H11O2熱分解とHO2生成の速度定数
c-C6H11は,Cl2の光分解(355nm)で生成したCl原子とシクロヘキサン(c-C6H12)の反応により生成させた.分子総密度[M]=1.2×1018 / molecule cm-3一定の条件において,[O2]>>[c-C6H11]とし,平衡濃度がc-C6H11O2に十分偏った条件において,c-C6H11O2をUV吸収法により観測し,熱分解速度定数を550,600,650,700Kで測定した.反応速度のアレニウスプロットより,以下のアレニウス式を得た.
k = 1.10×109exp(-19.7±1.7 kcal mol-1/RT) / s-1
また,近赤外周波数変調分光法により観測されたHO2のプロファイルより,HO2生成の速度定数を各温度に対して解析し,同様に求めたアレニウス式を以下に示す.
k = 3.58×1010exp(-22.1±4.2 kcal mol-1/RT) / s-1
観測されたHO2がc-C6H11O2の熱分解反応より生成する直接的な生成物であるとした場合,c-C6H11O2の熱分解速度定数とHO2生成速度定数は一致するが,HO2を観測して得られた反応速度は,c-C6H11O2を観測して得られた速度よりも5-8倍程度,600-700Kの温度範囲で大きい.本研究においては,この差異に起因している反応を明らかにする事は出来なかったが,c-C6H11O2のUV吸収シグナルに熱分解生成物に起因すると考えられる吸収シグナルがオーバーラップしたことにより,c-C6H11O2の熱分解速度定数が過小評価されたことが一因であると考えられる.
OH収率の定量
また同実験条件において,OHを近赤外光周波数変調分光法により観測し,収率の測定を行った.OHは500-700Kにおいて本実験装置では検出されず,収率の上限値をφOH<0.03-0.05と求めた.本測定条件においては,c-C6H11O2の熱分解により生成するOHはc-C6H12とすぐさま反応するため観測は困難であり,検出可能なのは,c-C6H11+O2の反応よりダイレクトに生成するOHラジカルである.ここのでの観測結果では,c-C6H11+O2の反応より生成するOHラジカルの収率の上限値を評価したものである.
HO2収率の定量
HO2の収率を298-750Kの温度範囲で測定したところ,500K付近より増加し,700K付近において1.0に飽和する傾向が得られた(Fig.5).これは既に報告されているC2H5,C3H7,c-C5H9+O2より生成するHO2の収率と類似しているが,c-C6H11+O2は他の炭化水素ラジカルの反応と比べると,やや高温側にシフトする傾向が得られた.
また,625Kにおいて,圧力を変化させHO2を観測した
ところ,c-C6H11+O2より生成すると考えられる素早いHO2の生成と,c-C6H11O2の熱分解に起因すると考えられる緩やかなHO2の生成が観測された.これは,c-C6H11O2で安定化せずにHO2を生成するPromptな成分が,観測した圧力範囲において圧力依存性を有しているためであると考えられる.625Kにおいて観測されたPrompt HO2とHO2 Totalの収率の圧力依存性をFig.6に示す.Prompt HO2の収率が観測した圧力領域において変化することより,c-C6H11+O2の反応速度が[M]=0.21-1.17×1018 molecule cm-3において圧力依存性を示すと考えられる.
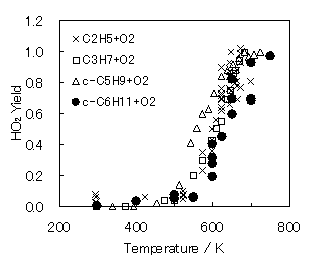
Fig.5 C2H5,C3H7,c-C5H9,c-C6H11+O2 より生成するHO2の収率の温度依存性
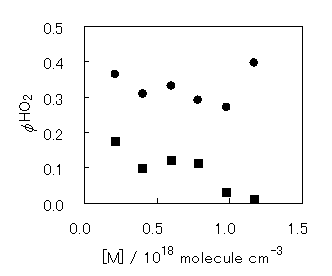
Fig.6 625KにおけるPrompt HO2(■)とTotal HO2(●)収率の圧力依存
3.3 c-C6H11+O2の低温酸化反応機構
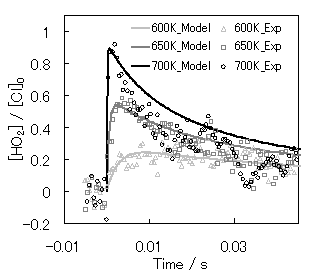
本実験において観測されたHO2のプロファイルを,測定したHO2の生成速度定数,HO2の収率,既に報告されているHO2の自己消費反応速度[8],HO2+c-C6H11O2の反応速度[9]を用いて反応モデルを提案し,本研究で観測されたHO2のプロファイルと比較した.HO2の立ち上がりに関しては,Promptな生成を考慮していないものの,良い一致を示し,減衰プロファイルはHO2の自己消費反応のみでは過小評価していることが明らかとなった(Fig.7).これらは,他の熱分解反応の影響によるものと考えられる.
3.4 まとめ
本研究では,c-C6H11O2とHO2の検出を行い,c-C6H11+O2の低温酸化反応に関して検討を行った.c-C6H11O2を観測して得られた熱分解速度定数と,HO2を観測して得られた生成速度定数が異なることより,c-C6H11O2を観測したUVシグナルには熱分解生成物のシグナルが影響し,反応速度定数を過小評価していると考えられる.また,OHの生成は観測されず,c-C6H11+O2の反応より直接生成するOHの収率は少ないと考えられる.また,HO2のTotalの収率は600Kより上昇し700K付近で1.0に飽和し,C2H5,C3H7同様の特性を示すことが確認された.また,低圧においてはc-C6H11+O2より生成するPromptな生成が観測され,圧力の低下に伴いPromptに生成するHO2の収率が増加する傾向が得られた.
Fig.7 反応モデルにより再現されたHO2プロファイル

4. 総括
本研究では,低温酸化過程で生成する重要な中間生成物と考えられるビノキシ型のラジカルに対して,置換基の付加位置により反応性がどのように変化するか解明した.また,実験的に未解明であったシクロヘキシルの低温酸化反応における反応経路と,反応機構に関して検討を行った.
参考文献
[1] M. Hassouna et al., J.
Phys. Chem. A, 110, 6667, 2006
[2] T. Oguchi et al., J.
Phys. Chem.A, 105, 378, 2001
[3]
K. Imrik et al., Phys. Chem. Chem. Phys, 6, 3958, 2004
[4]
R. Cox et al., Chem. Phys. Lett, 173, 206, 1990
[5] E. Clifford et al., J. Phys.
Chem. A, 104, 11549, 2000
[6] J. DeSain et al., J.
Phys. Chem. A, 105, 3205, 2001
[7] J. DeSain et al., J.
Phys. Chem. A, 105, 6646, 2001
[8] H. Hippler et al., J. Chem.
Phys., 93, 1755, 1990
[9] D. Rowley et al., J. Phys. Chem., 96, 4889, 1992